
Types of Rett syndrome
Last updated Sept. 22, 2025, by Marisa Wexler, MS
 Fact-checked by José Lopes, PhD
Fact-checked by José Lopes, PhD
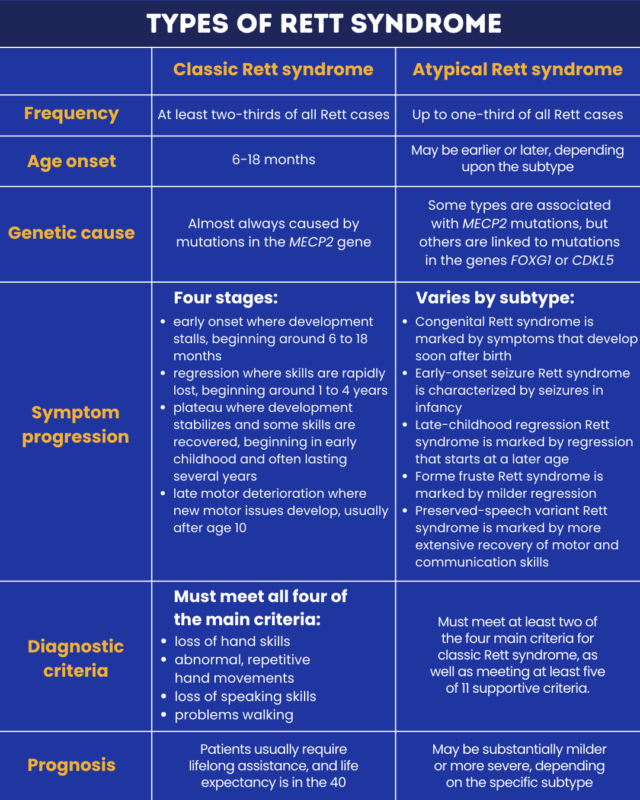
Rett syndrome is a rare genetic disorder marked by abnormalities in development that affect brain function. There are two main types of Rett syndrome: classic and atypical. Additionally, there are several subtypes of atypical Rett syndrome.
Although the different types share many of the same Rett syndrome symptoms, there are also notable differences in how the disease manifests. Factors that vary by type include the age at onset, the severity of symptoms, and the specific symptoms that tend to appear.
Classic Rett syndrome
Classic Rett syndrome is the most common form of the disease, accounting for at least two-thirds of all cases.
Almost all cases of this type are caused by mutations in the MECP2 gene. This gene is located on the sex-determining X chromosome, and consequently, classic Rett syndrome is much more common in girls, though in rare cases, it can also affect boys.
Babies with classic Rett syndrome will appear to develop normally over the first several months of life. Symptoms appear in early childhood, and then classic Rett syndrome progression tends to occur in four distinct stages:
- The early onset phase, usually occurring between 6 and 18 months of age, is when normal development stalls for a few months to a year.
- The rapid destructive phase, typically seen between ages 1 and 4, is when children will regress and lose skills they had previously acquired. This phase can occur over the course of weeks or months.
- The plateau phase is when development stabilizes, and some skills may be recovered. It typically occurs between ages 2 and 10, though this phase can last many years.
- The late motor deterioration phase, which usually occurs after age 10, is marked by the development of more substantial motor impairments while communication skills remain unchanged.
Although specific experiences can vary from person to person, all people with classic Rett syndrome will experience the pattern of normal early development, followed by regression, then stabilization or recovery. To be diagnosed with classic Rett syndrome, patients must have symptoms meeting all of the following four main criteria:
- a loss of purposeful hand skills that had previously been acquired
- a loss of spoken language abilities, such as losing the ability to say specific words or babbling less
- abnormal walking patterns or a complete inability to walk
- repetitive hand movements, such as squeezing or rubbing the hands together
Many people with classic Rett syndrome will also have other symptoms, which can range from seizures and behavioral disturbances to digestive problems and difficulty breathing.
Individuals of all ages with classic Rett syndrome will usually require help to go about their daily lives, though activities like eating or toileting may be done with minimal or no assistance. Most people with classic Rett survive into their 40s, but if symptoms are mild, a person with Rett syndrome may have a normal life expectancy.
Atypical variants of Rett syndrome
The terms atypical or variant Rett syndrome are used to refer to people who show many features of the disease but do not meet all the criteria for classic Rett syndrome. Reports show that atypical Rett syndrome accounts for as many as one-third of all Rett cases.
To qualify for a diagnosis of atypical Rett, individuals must show the usual pattern of normal development, followed by regression and then stabilization. Patients also must meet at least two of the four main criteria for classic Rett syndrome. Additionally, patients must meet at least five of 11 supportive criteria, which include slow growth, impaired sleep, and certain behavioral abnormalities.
Five specific subtypes of atypical Rett syndrome have been defined:
- congenital Rett syndrome
- early-onset seizure variant
- late-childhood regression form
- forme fruste variant
- preserved speech variant
Although all of these conditions have historically been regarded as Rett syndrome variants, today some of them are more typically viewed as separate conditions.
Congenital Rett syndrome
Congenital Rett syndrome, also known as the Rolando variant, is marked by symptoms that are similar to those of classic Rett syndrome. However, these symptoms have a distinctly earlier age at onset: Babies with congenital Rett syndrome usually start to show abnormalities soon after birth, instead of developing normally for several months as is the case in classic Rett syndrome.
Mutations in a gene called FOXG1 have been identified as a cause of congenital Rett syndrome. Because this gene is not located on a sex chromosome, congenital Rett syndrome affects both sexes at equal rates. Differences in how early symptoms occur and in the relative frequency in each sex have made physicians and researchers consider FOXG1 syndrome as a distinct entity from Rett syndrome.
Early-onset seizure Rett syndrome
Early-onset seizure Rett syndrome, also known as the Hanefeld variant, is characterized by seizures that begin in infancy, usually within the first few months after birth. This is distinct from classic Rett syndrome, where patients generally don’t experience seizures until later in childhood.
Rett syndrome in children with the early-onset seizure form also tends to manifest with developmental delays earlier in childhood than is typically seen with classic Rett syndrome. Sleep problems are also more common in the early-onset seizure variant, while an abnormal spine curvature is less common than in classic Rett syndrome.
The early-onset seizure variant of Rett syndrome is usually caused by mutations in a gene called CDKL5. Similar to congenital Rett syndrome, early-onset seizure Rett syndrome is today usually considered a distinct genetic condition referred to as CDKL5 deficiency disorder, or CDD.
Late-childhood regression Rett syndrome
The late-childhood regression form of the disease, also called late-onset Rett syndrome, is marked by a period of normal early development followed by development stopping and then regressing, similar to what’s seen in classic Rett syndrome. The key difference is that, in the late-childhood regression variant, the age at onset of symptoms is markedly older, and the loss of motor and language skills is more gradual.
As with classic Rett syndrome, the late-childhood regression form is typically associated with mutations in the MECP2 gene.
Forme fruste Rett syndrome
Forme fruste Rett syndrome is a relatively mild version of the disorder. People with forme fruste Rett syndrome tend to experience a slightly later age at onset, and the regression of developmental skills is usually slower and less pronounced. Compared with individuals with classic Rett syndrome, those with the forme fruste variant are more likely to retain motor and communication abilities, and are also more likely to have a normal head size.
Like classic Rett syndrome, the forme fruste variant is mainly associated with mutations in the MECP2 gene.
Preserved-speech variant Rett syndrome
The preserved-speech variant of Rett syndrome, also called the Zappella variant, starts off very similarly to classic Rett syndrome. However, in people with this variant, the age at onset of regression tends to be a bit later. Also, these patients experience a more extensive recovery of motor and speech skills than is seen in classic Rett syndrome. Autistic features may be noted with this subtype.
Like classic Rett syndrome, the preserved-speech variant is usually associated with a MECP2 gene mutation.
How doctors determine Rett type
A Rett syndrome diagnosis is made clinically — in other words, doctors diagnose the condition based on the specific symptoms that a patient experiences. Determining the type of Rett syndrome is likewise based on a patient’s symptoms.
According to current diagnostic criteria, a person may be diagnosed with classic Rett syndrome if:
- the individual shows a typical pattern of Rett development, with early normal development followed by regression and then stabilization or recovery
- the patient meets all four of the main criteria for classic Rett syndrome
- other conditions, like brain damage and dramatic early development abnormalities, have been ruled out
By the same criteria, atypical Rett syndrome may be diagnosed if a patient shows a Rett-like pattern of development as well as:
- meeting at least two of the four main criteria
- meeting at least five of the 11 supportive criteria
Distinguishing between the different subtypes of atypical Rett syndrome is likewise usually done by carefully evaluating a patient’s symptoms and clinical history to see which subtype is most appropriate. Genetic testing to look for mutations in MECP2, FOXG1, and/or CDKL5 may also be helpful for identifying the specific subtype.
Why knowing the type matters
There are a few reasons why it can be helpful to know the specific type of Rett syndrome that a person has. Knowing the type can help in:
- predicting how the disease is likely to evolve over time, which is important for making future plans
- guiding care choices, such as the use of Rett syndrome treatment options or assistive devices, which may be more appropriate for some types than others
- connecting with others in the community who are affected by the same type of Rett syndrome and may be dealing with similar issues
- determining eligibility for clinical trials testing investigational treatments
Rhett Syndrome News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.