

Causes of Rett syndrome
Last updated Sept. 12, 2025, by Marisa Wexler, MS
 Fact-checked by José Lopes, PhD
Fact-checked by José Lopes, PhD
Rett syndrome is a genetic disorder that mainly affects girls, causing symptoms such as developmental abnormalities, motor impairments, and difficulty with communication. The condition is usually not inherited from parents and instead develops spontaneously. Understanding how Rett syndrome happens can help inform families about the risk of having future children with this disorder.
What causes Rett syndrome?
Rett syndrome is caused by genetic mutations. The vast majority of cases — upwards of 95% — are caused by mutations in a gene called MECP2.
The MECP2 gene provides instructions to make a protein called MeCP2. Rett-causing mutations in the gene lead to a reduction in the amount of functional MeCP2 protein that is produced. Although the link between Rett syndrome genetics and the specific features of the disease aren’t fully understood, data indicate that the MeCP2 protein is vital for maintaining the health of nerve cells and other cells in the brain. In particular, MeCP2 is known to act as an activator or repressor of gene activity, helping to determine which genes are turned on and off. Reduction of MeCP2 functionality is thought to lead to abnormalities in gene activity, impaired communication between nerve cells, and issues with brain development, which ultimately cause Rett symptoms.
Although MECP2 mutations account for nearly all cases of Rett syndrome, not everyone with Rett has a mutation in this gene. A few other genes have also been linked to Rett syndrome, namely CDKL5 and FOXG1. Nearly 30 different types of disease-causing mutations have been identified in Rett syndrome genetic research.
Why Rett syndrome mainly affects girls
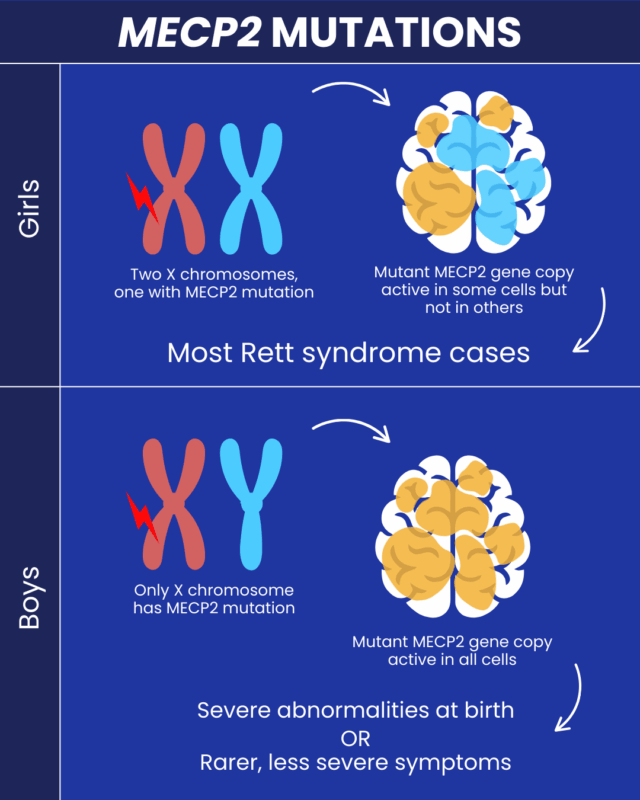
Rett syndrome mainly affects girls because the MECP2 gene is located on the X chromosome, which is one of the two sex-determining chromosomes. Although there are exceptions, generally biological females have two X chromosomes while biological males have one X and one Y. This means that most girls get two copies of the MECP2 gene, while most boys get only one.
In cells with two X chromosomes, only one of the chromosomes will be active while the other is inactivated. So, if a girl has one mutated MECP2 gene copy but a second healthy one, the mutated gene will be active only in some of her cells, while the healthy gene will be active in the rest of her cells. As a result, some cells will acquire disease-driving damage, but others will not.
By contrast, as they have only one X chromosome, males do not have a healthy gene copy to compensate for the MECP2 mutation. As a consequence, MECP2 mutations in boys tend to lead to a much more severe clinical picture. Boys with MECP2 mutations usually develop a condition called severe neonatal-onset encephalopathy, which is marked by profound abnormalities at birth. Of note, a period of normal development in the first months of life is typical in Rett, followed by regression later on.
Boys suspected of having Rett or otherwise showing MECP2 mutation symptoms may also be categorized into three further groups:
- they have mutations that are less severe than in other forms of Rett
- they have what is called genetic mosaicism, where the MECP2 gene is mutated in some cells but healthy in other cells
- they have Klinefelter’s syndrome, where the person inherits three sex chromosomes, two X and one Y.
How MECP2 mutations occur
In more than 99% of cases, MECP2 mutations that are the genetic cause of Rett syndrome develop in a de novo fashion. This means that a spontaneous mutation arises in sperm or egg cells, or during the growth of cells in early embryonic development. In these cases, the mutation is not present in either biological parent, so if those parents choose to have another child, the odds of having a second child with Rett syndrome are extremely low.
That said, MECP2 mutations may be inherited in some cases. Specifically, two situations can lead to Rett syndrome mutation inheritance:
- germline mosaicism, where a parent’s sperm or egg cells carry a MECP2 mutation that may be passed on to biological children, but the mutation is not present in the parent’s other cells, which means they don’t have Rett syndrome
- a mother carries a MECP2 mutation, but by random chance the X chromosome carrying the mutation is inactivated in almost all of her cells, so she doesn’t have Rett syndrome but still has a 50% chance of passing the mutation to her biological children.
Genetic testing and genetic counseling for Rett syndrome can help inform prospective parents about the risk of having biological children with the condition.
Other genes that can play a role
In addition to MECP2, mutations in two other genes have been identified as atypical Rett syndrome causes.
- FOXG1 mutations have been linked to congenital Rett syndrome, which is marked by developmental delay and loss of muscle tone that occur in the first months of life. The gene is not located on the X chromosome, so Rett syndrome associated with FOXG1 mutations is equally common in boys and girls.
- CDKL5 mutations have been associated with the early-onset seizure variant of Rett syndrome, or the Hanefeld variant, which is marked by seizures that develop in early infancy. The CDKL5 gene is located on the X chromosome like the MECP2 gene, so CDKL5-associated Rett syndrome is also more common in girls.
Although disorders caused by mutations in CDKL5 and FOXG1 have historically been considered forms of Rett syndrome, some experts are now considering these conditions to be distinct diseases.
Myths about the causes of Rett syndrome
Several misconceptions are common about the causes of Rett syndrome.
- Myth: Rett syndrome only affects girls
Fact: Rett syndrome usually affects girls, but it can also affect boys. - Myth: Everyone with a MECP2 gene mutation will develop Rett syndrome
Fact: Although many people with MECP2 mutations will develop Rett syndrome, some people who carry a mutation in this gene will never develop any symptoms of the syndrome. - Myth: Vaccines cause Rett syndrome
Fact: Rett syndrome is caused by genetic mutations that are present from birth. No credible evidence shows vaccines cause Rett syndrome or any other neurodevelopmental disorder. - Myth: Rett syndrome is always inherited
Fact: In the vast majority of cases, Rett syndrome is caused by de novo mutations, which arise at random in sperm or egg cells, or during embryonic development. This means that the mutation is not passed down from parents and that it is very unlikely that multiple children in the same family will have the condition. That said, Rett can be inherited in rare instances. Genetic testing and counseling can help inform prospective parents about the risk of having biological children with Rett. - Myth: Rett syndrome is a form of autism
Fact: Rett syndrome is not classified as an autism spectrum disorder. However, people with Rett may show autistic features, particularly during the regression stage, such as loss of social interaction. Differences between the two conditions include the loss of hand skills and issues with breathing in Rett, but not in autism. Also, unlike Rett, autism is diagnosed more often in boys.
Rett Syndrome News is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
Related articles
-
-
 Discussion
Discussion
-
 Discussion
Discussion
-
-
Discussion
-
